










高通量虚拟筛选服务基于计算机模拟方法,通过快速筛选大量化合物库以寻找潜在的活性分子。Neorabio采用高效对接算法和并行计算技术,能够在短时间内完成数十万甚至数百万化合物的筛选,极大地提升早期药物发现的效率。该服务适用于先导化合物的发现、药物靶标筛选和成药性优化,广泛应用于制药公司和科研机构的药物开发项目中。Neorabio的高通量虚拟筛选帮助客户大幅提高研发效率和精准度,加速药物候选分子的筛选过程。
服务流程
[提交结构] → [结构预处理] → [参数设置] → [对接运行] → [结果分析] → [报告交付]
应用场景
基于靶点结构或配体信息的大规模化合物筛选
快速筛选海量化合物库,发现潜在活性分子
结合打分与筛选策略,优选候选分子
辅助靶点成药性评估及机制研究
为后续实验筛选提供高质量候选分子
支持结构优化与药效团建模的后续工作
节约实验成本,提高筛选效率
技术优势
我们拥有高效的计算平台,支持大规模化合物库的快速筛选
结合多种筛选算法,包括基于结构的分子对接和基于配体的药效团建模,提升筛选准确性
完善的蛋白质结构和小分子预处理流程,确保数据质量和对接效果
支持柔性对接和多结合位点筛选,适应复杂靶点的筛选需求
提供筛选结果的多维度分析与可视化,帮助客户精准选取潜在活性分子
具备丰富的定制化方案,满足不同科研和产业项目的个性化需求
经验丰富的专业团队,结合生物学背景,助力靶点验证与药物设计
经典案例
计算机辅助结构虚拟筛选的药物重定位方法鉴定SARS-CoV-2病毒侵入抑制剂
基于药效团的虚拟筛选用于发现新型抗包虫病化合物
研究背景:
基于新冠病毒 SARS-CoV-2 的迅速传播以及它对全球公共健康的严重威胁。SARS-CoV-2 通过其表面刺突糖蛋白(S-蛋白)与人类细胞受体 ACE2 结合,从而进入宿主细胞。这一病毒与宿主的特异性分子相互作用提供了一个潜在的治疗靶点。本研究通过计算机虚拟筛选,筛查了多个 FDA 批准的药物库,寻找可以干扰 SARS-CoV-2 和 ACE2 之间相互作用的候选分子,并对其结合能量和结合模式进行了详细分析,进而筛选出可能抑制的药物。
方案设计:
靶点选择,同源建模,分子库筛选,分子对接,分子动力学模拟,候选分子验证
主要结果:
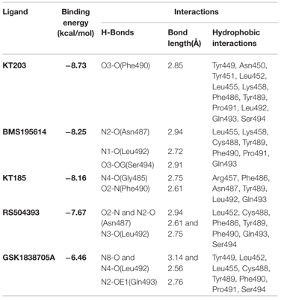
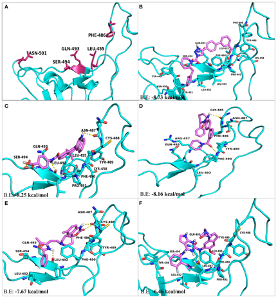
1、ACE2受体与FDA化合物库筛选、优势化合物和ACE2分子对接
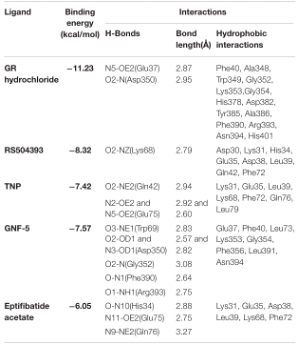
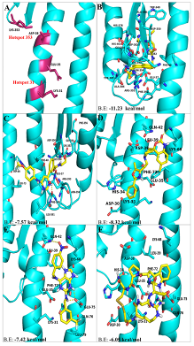
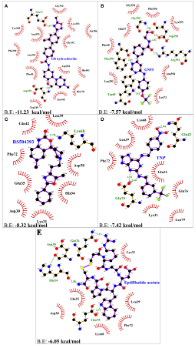
图A筛选化合物亲和力排序 图B 3D相互作用图 图C 2D相互作用图 图D 动力学模拟结合稳定性
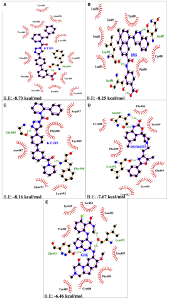
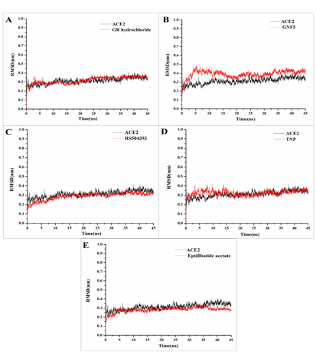
图A筛选化合物亲和力排序 图B 3D相互作用图 图C 2D相互作用图 图D 动力学模拟结合稳定性
结果显示,RS504393不仅能够结合ACE2受体的病毒结合位点,还能够结合病毒S蛋白的受体结合位点,因此潜在抑制病毒入侵过程有效地帮助控制SARS-CoV-2快速传播的效果。
参考文献
Choudhary S, Malik YS, Tomar S. Identification of SARS-CoV-2 Cell Entry Inhibitors by Drug Repurposing Using in silico Structure-Based Virtual Screening Approach. Front Immunol. 2020 Jul 10;11:1664. doi: 10.3389/fimmu.2020.01664. PMID: 32754161; PMCID: PMC7365927.
研究背景:
棘球蚴病(包虫病)是一种严重的人畜共患寄生虫病,对人类健康和畜牧业造成重大影响。然而,临床使用的药物(苯并咪唑)治愈率低,急需替代药物。目前对棘球蚴病的药物筛选主要以表型为主,对活性化合物的识别效率很低。本研究利用活性氨基醇结构生成的药效团模型进行了虚拟筛选以发现具有抗棘球蚴活性的新化合物。
方案设计:
生成并验证药效团模型;基于药效团模型的虚拟筛选;体外药物筛选;细胞毒性测试;
主要结果:
1.药效团模型生成、验证和虚拟筛选
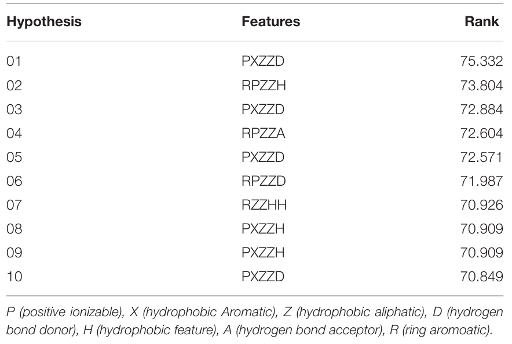
生成10个HipHop模型后,用活性和非活性氨基醇组成的测试集来选择最佳模型,其中药效团模型HipHop-Hypo03最佳。该药效团包括一个PI基团、一个HAR基团、两个HAL基团和一个HBD基团。
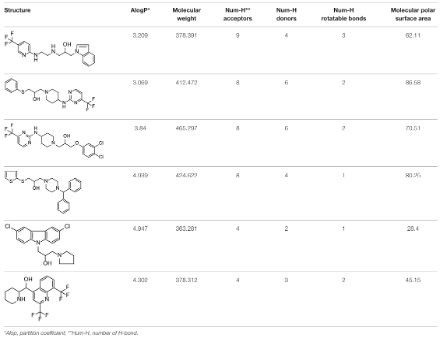
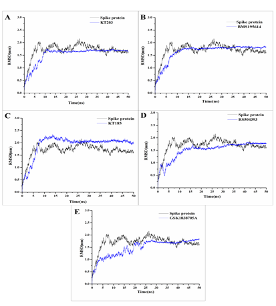
化合物训练集分子基本性质 模型质量评估
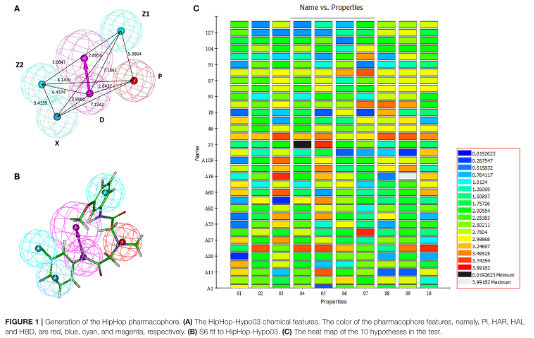
产生HipHOP药效团 寄生虫形态结构变化
2.评价候选化合物
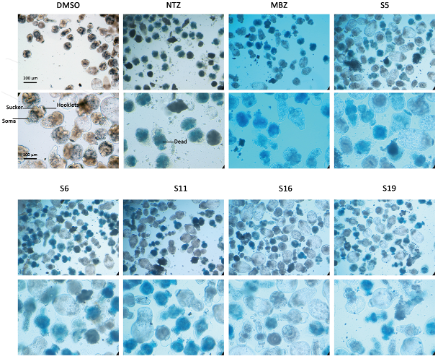
对E. multilocularis PSCs的体外活性及其细胞毒性10种化合物致使寄生虫在3天内100%死亡,它们的LC50值为16.99-20.45 μM。暴露于这些化合物会导致寄生虫的形态损伤(图2) 。此外,化合物S6、 S11. S16和S19的细胞毒性低,且S5、 S6、 S11、 S16、 S19、 S28能抑制癌细胞(A172细胞)的增殖。
参考文献
Liu C, Yin J, Yao J, Xu Z, Tao Y, Zhang H. Pharmacophore-Based Virtual Screening Toward the Discovery of Novel Anti-echinococcal Compounds. Front Cell Infect Microbiol. 2020 Mar 20;10:118. doi: 10.3389/fcimb.2020.00118. PMID: 32266168; PMCID: PMC7098963.
交付内容
虚拟筛选亲和力打分排序表及指定化合物相互作用分析
交付内容